

鐵死亡及其研究進展
細胞程序性死亡是由基因決定的受到分子機制精密調控的細胞主動,、有序的死亡方式,與生命體穩(wěn)態(tài)的維持和疾病的發(fā)生密切相關,,主要包括焦亡,、壞死和自噬等形式。鐵死亡是近幾年發(fā)現(xiàn)的一種新的細胞程序性死亡方式,,作為一種新的細胞死亡方式已成為科研人員瞄準的新方向,。鐵死亡這一概念最早在2012年由Dr. Dixon [1] 提出,,它是一種鐵依賴性的脂質過氧化,、活性氧自由基大量累積所致的細胞死亡模式,受到多種細胞代謝途徑的調控,,其中包括氧化還原穩(wěn)態(tài),、鐵代謝、線粒體活性和氨基酸,、脂質,、糖的代謝,以及各種與疾病相關的信號途徑,。研究表明,,鐵死亡與器官損傷、神經(jīng)退行性疾病,、心血管疾病,、炎癥性疾病、癌癥相關。特別是具有多重藥物抗藥性的癌細胞,,尤其是處于間充質狀態(tài)且易于轉移的腫瘤細胞,,非常容易發(fā)生鐵死亡。因此,,通過誘導和抑制鐵死亡對其進行藥理調節(jié),,在治療耐藥性癌癥、缺血性器官損傷和其他與脂質過氧化密切相關的退行性疾病上具有巨大的潛力,。

圖1 細胞死亡的分類
鐵死亡的生化特點: 鐵死亡又名鐵壞死,,是一種與活性氧(reactive oxygen species, ROS)聚集有關的細胞死亡形式,其本質為鐵蓄積和脂質過氧化,。細胞抗氧化體系代謝異常時,,Fe2+蓄積能夠介導芬頓(Fenton)反應產(chǎn)生過量ROS(尤其是羥自由基),ROS與細胞膜上的多不飽和脂肪酸(polyunsaturated fatty acid, PUFA)發(fā)生過氧化反應,,導致脂質雙分子層穩(wěn)定性遭到破壞,,細胞膜解體,進而促進細胞鐵死亡[1] ,。

圖3 鐵死亡的主要途徑
2.鐵死亡主要途徑
鐵死亡可通過外源性或內源性途徑誘發(fā),。
2.1外源性 (轉運蛋白依賴) 途徑
2.1.1氨基酸代謝—抑制胱氨酸/谷氨酸轉運蛋白 [5]
System xc-是細胞內重要的抗氧化體系,該系統(tǒng)由2個亞基組成,,SLC7A11和SLC3A2,。SLC7A11負責主要的轉運活性,對胱氨酸和谷氨酸有高度特異性,,而 SLC3A2則作為伴侶蛋白,。System xc-以1:1的比例用胞內谷氨酸來換取胞外的胱氨酸(Cys2),胱氨酸在谷氨酸半胱氨酸連接酶(GCL)和谷胱甘肽合成酶(GSS)的催化作用下合成谷胱甘肽(GSH)(GSH是膜脂修復酶-谷胱甘肽過氧化物酶(GPX4)的還原性輔因子),。抑制 System xc- 的活性會抑制胱氨酸的吸收,,影響GSH的合成,繼而導致膜脂修復酶GPX4活性降低,,細胞抗氧化能力降低,,從而促進鐵死亡。
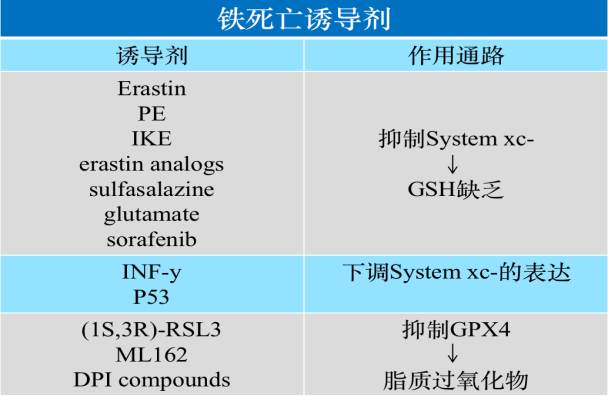
表2 目前已知的部分鐵死亡誘導劑及作用通路
2.1.2鐵代謝—轉運蛋白與鐵“超載” [6-7]
鐵具有Fe2+和Fe3+兩種氧化態(tài),。食物中Fe3+在腸道被還原為Fe2+后被小腸上皮細胞吸收,。Fe2+在膜鐵轉運蛋白的作用下被運輸至細胞外并被氧化為Fe3+,與轉鐵蛋白(transferrin, TF)(血清轉鐵蛋白或乳鐵蛋白)結合形成TF-Fe3+復合物,,經(jīng)血液循環(huán)運輸至各組織器官,。Fe3+與細胞膜上的轉鐵蛋白受體(TFRC)結合進入細胞內后被金屬還原酶STEAP3還原為Fe2+,然后通過二價金屬轉運蛋白1將Fe2+釋放到胞質的動態(tài)鐵池中,。正常生理狀態(tài)下,,動態(tài)鐵池可以維持鐵平衡,。
病理情況下,Fe 2+在細胞內集聚,,發(fā)生哈伯-韋斯反應(Haber-Weiss)與Fenton反應,,產(chǎn)生大量ROS,與細胞膜上的多不飽和脂肪酸PUFA發(fā)生一系列過氧化反應,,生成脂質過氧化物,,破壞細胞膜結構并引起細胞鐵死亡。同時Fe2+作為多種代謝鐵的積累是鐵死亡過程中啟動膜氧化的關鍵信號之一,;TF通過TFRG介導鐵攝取,,FTH1/FTL (鐵蛋白組件)通過自噬降解可以增加鐵的水平并促進鐵死亡。
SLC40A1介導的鐵外流和外泌體介導的鐵蛋白輸出會抑制鐵死亡,。
2.2內源性 (酶調控) 途徑
2.2.1脂質代謝—多不飽和脂肪酸PUFAs過氧化[8-9]
鐵死亡的中心環(huán)節(jié)是鐵依賴的脂質氧化代謝失調,,多不飽和脂肪酸(PUFAs)是鐵死亡中脂質過氧化物積累的關鍵物質。 正常情況下,,PUFAs 是脂質代謝的重要底物,,含有雙烯丙基氫原子,尤其是花生四烯酸(arachidonoyl, AA)和腎上腺素酸(adrenoyl, AdA),,易與ROS發(fā)生反應,,引起脂質過氧化。當長鏈脂酰輔酶A合成酶4(ACSL4)催化游離的AA或AdA與輔酶A(coenzyme A, CoA)結合形成衍生物AA-CoA或AdA-CoA,,然后被溶血卵磷脂?;D移酶3(LPCAT3)酯化為膜磷脂酰乙醇胺(PEs),并經(jīng)過脂氧合酶(ALOXs)或細胞色素P450氧化還原酶(POR)氧化后,,就形成了有害的脂質過氧化產(chǎn)物,,誘導細胞鐵死亡。
乙酰輔酶A羧化酶(ACAC)介導的脂肪酸合成,,脂質吞噬(Lipophagy)介導的脂肪酸釋放,,可誘導細胞內游離脂肪酸的積累,促進鐵死亡,。
2.2.2其他代謝途徑
CoQ10能被鐵死亡抑制蛋白1(ferroptosis suppressor protein 1, FSP1)還原來阻止脂質氧化進而抑制鐵死亡,。
NADPH能參與GSH-GPX4抗氧化系統(tǒng)的循環(huán),,其大量消耗將限制GSH-GPX4的抗氧化功能,,誘導鐵死亡。
硒是維持GPX4活性的一種必需的微量營養(yǎng)素,,通過協(xié)同激活轉錄因子TFAP2c和Sp1調節(jié)GPX4的豐度和活性,,在一定程度上抑制鐵死亡以保護神經(jīng)元。
NFE2L2可通過反式激活的方式調控包括鐵代謝,、GSH代謝以及抗ROS過程中的基因的表達進而限制鐵死亡過程中的氧化損傷,。
Vitamin E通過抑制ALOXs活性,、減少脂質過氧化物的產(chǎn)生,從而抑制鐵死亡,。
p53可以通過下調Xc-系統(tǒng)組分SLC7A11的表達,,抑制胱氨酸的攝取,從而誘導細胞鐵死亡,;同時p53可以抑制二肽基肽酶-4(dipeptidyl peptidase-4, DPP4)的活性,,阻斷Erastin誘導的鐵死亡。
NRF2是維持細胞內氧化還原穩(wěn)態(tài)的重要調控因子,,通過p62-Keap1-NRF2途徑上調參與鐵和ROS代謝的多種基因(NQO1,、HO1和FTH1)的表達,抑制細胞鐵死亡,。
3鐵死亡在疾病治療中的應用
鐵死亡與癌癥,、衰老、神經(jīng)退行性疾病,、心肌病,、中風、腦出血創(chuàng)傷性腦損傷,、缺血
再灌注損傷和腎臟變性相關的病理細胞死亡有關,。
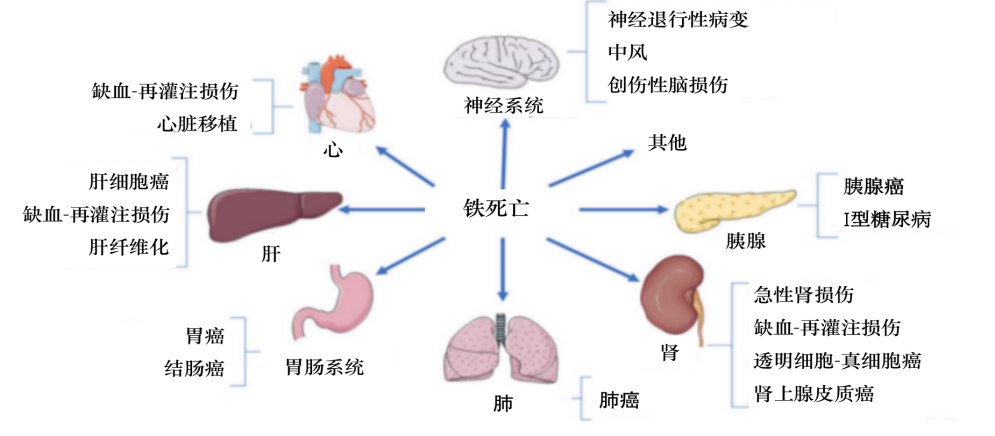
圖5 鐵死亡相關疾病
3.1 癌癥
研究表明許多癌癥相關基因和信號通路都能調控鐵死亡,誘導癌細胞鐵死亡對治療癌癥意義重大,。對凋亡和常規(guī)癌癥療法抵抗的間充質干細胞和去分化的癌細胞,,以及被稱為“持續(xù)細胞”的癌細胞都對鐵死亡誘導高度敏感。與正常細胞相比,,癌細胞代謝比較旺盛,,線粒體出現(xiàn)功能障礙,ROS積累較多,,增加了癌細胞對鐵死亡的敏感性,。
卵巢癌:實驗證實在卵巢癌中可通過TAZ-ANGPTL4-NOX2信號軸激活鐵死亡抑制卵巢癌細胞的增殖。
結,、直腸癌:細胞紅蛋白(cytoglob, CYGB)是ROS的細胞調節(jié)劑,,也是一種腫瘤抑制劑,能夠促進細胞膜上的脂質過氧化進而抑制結直腸癌細胞的生長,,過表達CYGB的結直腸癌細胞對RSL3和Erastin誘導的鐵死亡更敏感,,敲除Yes相關蛋白1基因YAP1可通過減輕ROS的產(chǎn)生降低結、直腸癌細胞對鐵死亡的敏感性,。
另外,,癌癥的治療可以通過靶向氨基酸、脂質,、鐵代謝相關信號通路進行,。
3.2 衰老和神經(jīng)退行性疾病
在衰老和退行性疾病如帕金森病(Parkinson's disease, PD),、亨廷頓病和阿爾茨海默病(Alzheimer's disease, AD)的患者中檢測到鐵水平升高,其原因可能是隨著年齡的增長,,鐵的代謝速率不斷降低,,導致機體鐵過量和過氧化損傷,最終引起細胞死亡,。
多種證據(jù)表明,,GSH-GPX4抗氧化通路在AD和PD患者中均出現(xiàn)明顯異常,使用抗氧化藥物可以緩解AD與PD病理癥狀,。
研究顯示,,使用鐵死亡抑制劑能夠保護帕金森病模型小鼠免受過氧化損傷。
大鼠海馬切片培養(yǎng)的離體實驗表明,,鐵死亡抑制劑ferrostatin-1可阻斷谷氨酸誘導的神經(jīng)元興奮毒性細胞死亡,。
遺傳學研究一致證實,小鼠條件性缺失Gpx4會引起類似神經(jīng)退行性病變的癥狀,。
3.3缺血再灌注損傷
缺血再灌注損傷是一種無菌性炎癥,。器官或組織缺血一段時間后重新獲得血液供應,但缺血性損傷不但沒有減輕反而進一步加重,,甚至出現(xiàn)更嚴重的細胞代謝功能障礙或結構破壞,。在腦缺血-再灌注損傷中鐵蓄積和脂質過氧化等現(xiàn)象與鐵死亡一致,且這一現(xiàn)象可以被鐵螯合劑或抗氧化劑抑制,。
在小鼠心臟缺血-再灌注損傷模型中,,在再灌注開始時給予抑制鐵死亡的小分子化合物Liproxstatin-1能夠增加線粒體內GPX4的含量,降低線粒體內ROS的產(chǎn)生,,降低缺血性梗死面積并改善線粒體結構和功能,。
在糖尿病大鼠心臟缺血-再灌注模型中發(fā)現(xiàn), ROS與內質網(wǎng)應激相互聯(lián)系是引發(fā)鐵死亡的重要因素,,其能夠進一步造成心臟缺血-再灌注損傷,。
在缺血-再灌注損傷小鼠模型、Gpx4全身敲除小鼠和葉酸誘導急性腎損傷小鼠模型中,,鐵死亡抑制劑已被證明可以減輕腎小管細胞死亡和急性腎功能衰竭,。
肝細胞特異性Gpx4敲除小鼠出生不久即會死亡,但高水平的維生素E飲食可以彌補肝臟中GPX4的缺乏,,使小鼠存活,。
此外Liproxstatin-1(一種強效鐵死亡抑制劑)能保護肝實質組織免受缺血-再灌注損傷。
3.4心腦血管疾病
發(fā)生腦卒中時,,胞內脂質過氧化物及 Fe2+水平升高,,而使用鐵死亡抑制劑上調 GSH、GPX4或 System xc-水平均可減輕腦部損傷,,這表明鐵死亡介導了腦卒中,。近年研究進一步發(fā)現(xiàn),機體鐵過載會降低動脈斑塊的穩(wěn)定性,,使用鐵螯合劑可縮小動脈斑塊面積,,減輕動脈粥樣硬化的發(fā)展。心肌細胞凋亡是心力衰竭的重要病理變化,,但研究表明心力衰竭過程中除細胞凋亡外還有鐵死亡的參與,,且兩者獨立并存。
4細胞鐵死亡的檢測
4.1形態(tài)學檢測
超微形態(tài)學特征顯示細胞膜斷裂和出泡,,線粒體變小,、膜密度增高、線粒體脊減少或消失,、線粒體外膜斷裂,,細胞核大小正常、但缺乏染色質凝聚,。電鏡下表現(xiàn)為細胞內線粒體變小及雙層膜密度增高,。
4.2生物學特征
*鐵死亡的金指標:脂質過氧化物(Lipid peroxidation)
鐵和活性氧(ROS)聚集,激活絲裂原活化蛋白激酶(mitogen-activatedprotein kinase,,MAPK)系統(tǒng),,通過降低胱氨酸的攝取、耗竭谷胱甘肽,,抑制ystem Xc-和增加還原型酰腺嘌呤二核苷酸磷酸氧化酶,,釋放花生四烯酸等介質。細胞內活性氧和脂質活性氧通過流式細胞術使用DCFH-DA,、C11-BODIPY 和Liperfluo檢測,。
在細胞培養(yǎng)基中添加藥物后,脂質過氧化物染色(Liperfluo)檢測Lipid peroxidation,,如為陽性結果,,證明鐵死亡發(fā)生;若在上述添加藥物的細胞培養(yǎng)基中添加鐵死亡抑制劑后,,檢測Lipid peroxidation為陰性結果,,證明藥物誘導的一定是鐵死亡,而非其他死亡方式,。
C11-BODIPY對自由基非常敏感,,但不和脂質過氧化物反應,Liperfluo是目前唯一可以特異性檢測脂質過氧化物的化合物,。
4.3鐵水平檢測
可以使用PGSK探針,流式細胞術或共聚焦顯微鏡檢測細胞內鐵含量的細胞膜透性染料,;或者使用Iron Assay Kit(試劑盒)檢測細胞,、組織中的鐵水平,。
實驗原理:通常利用鐵離子氧化狀態(tài)的過渡性改變來進行各種化學反應。通過加入酸性緩沖液來釋放鐵離子,,樣品中的Fe2+可直接測定,,或通過還原反應來測定鐵元素(Fe2+和Fe3+)總含量。反應釋放出的鐵元素與生色團反應,,生成與樣品中的鐵成正比的比色(593 nm)產(chǎn)物,。
4.4細胞活性檢測
可以使用MTT/CCK8法檢測細胞活性。
4.5線粒體膜電位檢測
可以使用TMRE熒光染料檢測,,TMRE能夠特異性標記有活性的線粒體,,是一種細胞膜可穿透性的,正電荷的,,橘紅色熒光染料,。當線粒體膜電位降低時,熒光減弱,。
實驗原理:TMRE (tetramethylrhodamine, ethyl ester),,中文名稱叫做四甲基羅丹明乙酯,CAS號為115532-52-0,,分子式為C26H27CIN2O7,,分子量為515。TMRE是一種可滲透細胞膜的橘紅色陽離子熒光探針,,可在完整的線粒體中聚集,,而去極化或非活躍性線粒體膜電位降低,導致TMRE積聚減少,。檢測時TMRE的最大激發(fā)波長為550nm,,最大發(fā)射波長為575nm。正常狀態(tài)下的線粒體內部存在大量的負電荷,,作為陽離子探針的TMRE進入細胞內后可聚集在線粒體基質中,,可發(fā)出明亮的橘紅色熒光;當細胞發(fā)生凋亡時,,線粒體膜電位丟失,,線粒體通透性轉換孔(MPTP)持續(xù)開放,TMRE被釋放到細胞質中,,線粒體內橘紅色熒光強度明顯下降,。可用熒光顯微鏡,、流式細胞儀、熒光酶標儀等儀器檢測細胞,通過熒光信號的強弱來確定線粒體膜電位的變化和凋亡或壞死的發(fā)生,。
4.6 qRT-PCR/Western blot檢測
檢測細胞內與鐵死亡相關的因子的變化,,例如COX-2,ACSL4,,PTGS2,,NOX1,,GPX4和FTH1等,,其中COX-2,ACSL4,,PTGS2和NOX1在鐵死亡細胞中表達上調,;GPX4和FTH1在鐵死亡細胞中表達下調。
參考文獻:
[1]Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell, 2012, 149(5): 1060-1072.
[2]陳璐瑤,饒小珍.鐵死亡的發(fā)生機制及相關疾病研究進展[J].生物學教學,2022,47(02):2-5.
[3]Jiang X, Stockwell BR, Conrad M.Ferroptosis: mechanisms, biology and role in disease.Nat Rev Mol Cell Biol, 2021,22(4):266-282.
[4]Daolin Tang, Xin Chen, Rui Kang, Guido Kroemer. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021 Jan 29.
[5]Piani D, Fontana A. Involvement of the cystine transport system xc- in the Macrophage
-induced glutamate-dependent cytotoxicity to neurons. J Immunol, 1994, 152(7): 3578-3585.
[6] Outten FW, Theil EC. Iron-based redox switches in biology. Antioxid Redox Signal, 2009, 11(5): 1029-1046
[7] Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cel, 2015, 59(2): 298-308
[8]Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA, 2016, 113(34): E4966-E4975
[9]Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol, 2017, 13(1): 91-98
| contact us Address: Room 402, West Block, Ganghongji Business Building, Nanshan District, Shenzhen 1107, Building 1, Hengtaiyu Building, Tongtong Road, Fenghuang Street, Guangming District, Shenzhen 11th Floor, Second Training Building, Guangdong Light Industry Vocational and Technical College, No. 152 Xingang West Road, Haizhu District, Guangzhou City Tel:0755-86325431 facsimile:0755-86325431 Service email:[email protected] |

